Sign up to receive updates about new posts on this collaborative blog.


Oncology leaders will be hearing the term “Oncology Data Specialist” a lot in the coming years. But what does it mean – and how...
Topics: cancer research, oncology informatics

In the landscape of modern healthcare, the seamless flow of accurate and comprehensive information is not just a convenience but...
Topics: cancer research, oncology informatics
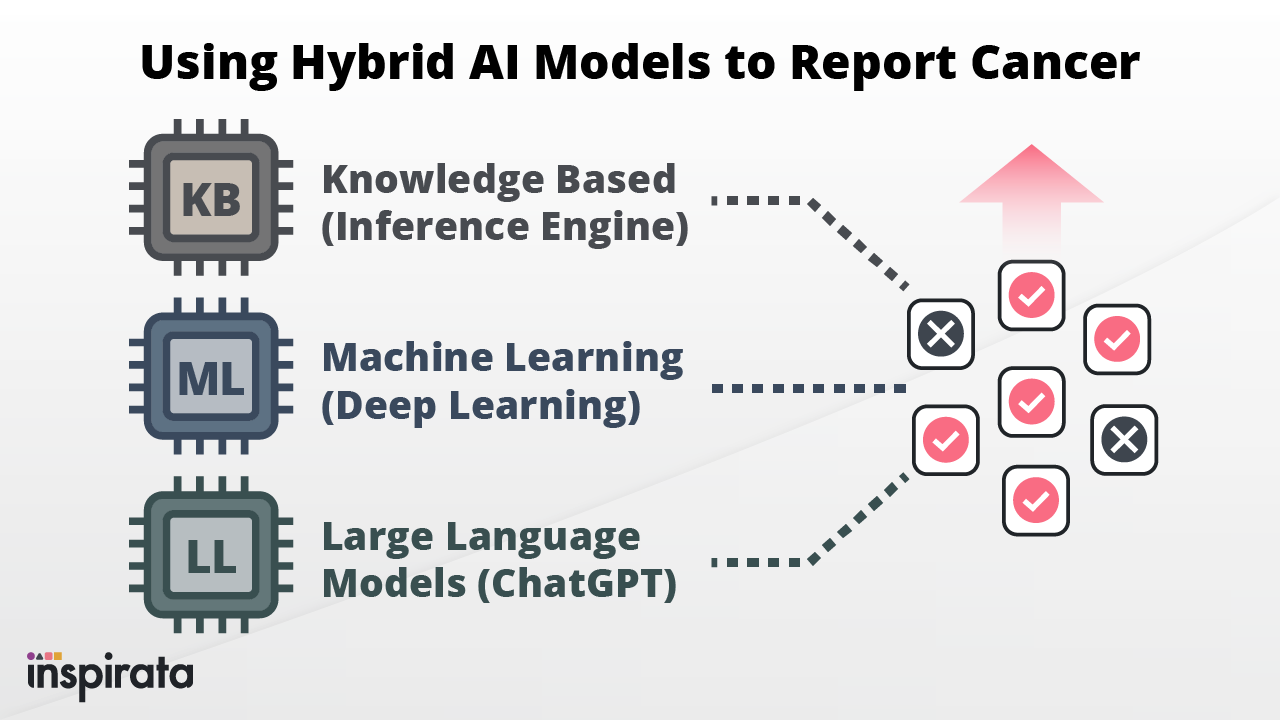
In recent years, various sectors of the healthcare industry have seen a significant increase in the use of Artificial...
Topics: artificial intelligence, cancer registry, AI/NLP

Responsible AI is far more than just a talking point. It's a core component of how forward-thinking companies like Inspirata are...
Topics: artificial intelligence, AI/NLP, oncology informatics

Every year on September 24th, the healthcare and research communities come together to acknowledge World Cancer Research Day....
Topics: cancer research

At Inspirata, we believe that innovation is a never-ending journey. We're committed to the ongoing process of identifying,...
Topics: cancer informatics, AI/NLP, quality control

It’s no secret that there has been a growing awareness of the importance of diversity, equity, and inclusion (DE&I) in clinical...
Topics: clinical trials, clinical studies, DI&E,

Clinical trial participants hail from all walks of life. They are young and old, representing myriad races and ethnicities, and...
Topics: artificial intelligence, clinical trials, clinical studies,

At Inspirata, we continuously strive to understand and address the unique needs of the cancer registry community. To gain insight...
Topics: cancer informatics, NLP, cancer registry

Achieving accrual targets for oncology clinical trials has always been a significant challenge, but more recently, it seems to be...
Topics: cancer informatics, clinical trials, clinical studies,

What can you do in a second or two of time? Most of us do not give it much thought, but in the right context, you may be...
Topics: ai, cancer informatics, NLP,

At Inspirata, we believe that using technology such as Artificial Intelligence (AI) and Natural Language Processing (NLP) to...
Topics: clinical trials, clinical studies, DI&E,
In a prior post, I endeavoured to introduce the importance of patient and public involvement (PPI) in the delivery of cancer...
Topics: ai, cancer informatics, clinical studies,

The instant my haematologist mentioned the word cancer, I stopped listening. Not purposely of course, I was in shock. Rather than...
Topics: cancer informatics, clinical studies, patient advocacy,

This week, April 5th – 9th, 2021, marks the 25th annual National Cancer Registrars Week (NCRW). This year’s theme, “Cancer...
Topics: cancer informatics, national cancer registrars week, cancer registry

At the end of 2020, I was delighted to take up the opportunity to join Inspirata as its business unit lead for cancer...
Topics: cancer informatics, cancer research, national cancer registrars week,

The astonishing success of artificial intelligence (AI) over the past few years in solving complex recognition problems has...
Topics: ai, artificial intelligence, cancer informatics,

During a recent virtual fireside chat, I met with industry experts Courtney Hudson, President and Founder of Carebox; and Cyntha...
Topics: NLP, clinical trials, cancer research,
Back in May 2020, at the early stage of the COVID-19 pandemic, the Inspirata team was very much focused on supporting the...
Topics: ai, cancer informatics, NLP

Passing information to the next generation is critically important.
Ten years ago, when my son was born, I had an epiphany. The...

We would like to hear from anyone willing to share their ideas. For those who are feeling adventurous and creative, we would like...





.jpg)





